FDA information on Expanded Access to Investigational Drugs/Biologics
For Physicians: How to Request Single Patient Expanded Access
FDA's Expanded Access Contact Information
21 CFR 312 Subpart I.
Expanded Access to Investigational Drugs for Treatment Use – Questions and Answers.
Keywords, Definitions and Resources
Refer to the expanded access categories and Title 21 of the Code of Federal Regulations (21 CFR) for more detailed information about expanded access request types.
21 CFR 312.310 Individual patients, including emergency use
21 CFR 312.315 Intermediate-size patient populations
21 CFR 312.320 Treatment IND or treatment protocol
Under FDA regulations (21 CFR 312.300), expanded access is the use of an investigational drug/biologic (referred to as “test article”) outside of a clinical trial for the diagnosis, monitoring, or treatment of a serious disease or condition when there is no satisfactory alternative therapy to treat the patient’s disease or condition. In contrast, participants in clinical trials/studies are considered human subjects whether they are patients or healthy volunteers. The main distinction between expanded access and the use of an investigational drug in the clinical trials covered under an IND, is that in most cases, traditional clinical trials are for the purpose of collecting safety and effectiveness data. The purpose of expanded access is to provide treatment. While expanded access is not considered a clinical investigation, FDA submission and IRB review are required.
Under the applicable criteria in 21 CFR 312.310(a), the physician must determine that the probable risk to the person from the investigational drug/biologic is not greater than the probable risk from the disease or condition; and the FDA must determine that the patient cannot obtain the investigational drug/biologic under another IND or protocol.
312.305 Requirements for ALL expanded access use:
(1) Patient(s) have a serious or immediately life-threatening disease or condition, and there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition;
(2) The potential patient benefit justifies the potential risks of the treatment use and those potential risks are not unreasonable in the context of the disease or condition to be treated; and
(3) Providing the investigational drug for the requested use will not interfere with the initiation, conduct, or completion of clinical investigations that could support marketing approval of the expanded access use or otherwise compromise the potential development of the expanded access use.
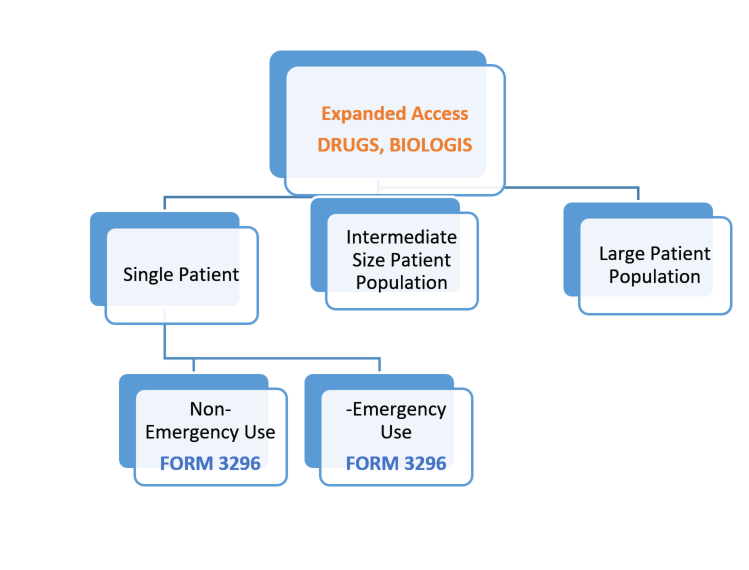
Under FDA’s current regulations, there are three categories of expanded access as shown in the diagram that follows. The primary difference in the three types of access is the number of patient(s) participating.
- Expanded access for individual patients, includes (1) emergency use (21 CFR 312.310) and (2) non-emergency use
- Expanded access for intermediate-size patient populations (generally smaller than those typical of a treatment IND or treatment protocol — a treatment protocol is submitted as a protocol to an existing IND by the sponsor of the existing IND) (21 CFR 312.315)
- Expanded access for wide-spread treatment use through a treatment IND or treatment protocol (designed for use in larger patient populations) (21 CFR 312.320)
For each category of expanded access, there are two (2) types of regulatory submissions to the FDA to obtain the IND:
(1) An expanded access protocol submitted as a protocol amendment to an EXISTING IND (i.e., an expanded access protocol) or
(2) A NEW IND submission, which is separate and distinct from any existing INDs and is intended only to make a drug/biologic available for treatment use (i.e., an expanded access IND)
For Emergency single patient treatment, you are NOT required to secure FDA and IRB review PRIOR to the use of the investigational treatment. Notification to the IRB must occur no later than 5 working days following administration of the test article.
For non-emergency single patient treatment, you ARE REQUIRED to notify the IRB PRIOR to the use of the test article and obtain FDA authorization before use.
Informed Consent: Informed consent must be sought from the subject or the subject’s legally authorized representative (LAR). The consent form should contain all the standard elements of informed consent. If the subject is unable to sign the consent, it may be signed by either a legal guardian or an attorney-in-fact with the authority to make health care decisions. If a person fulfilling those requirements is not available, review Virginia law allowing for the consent to be signed by a family member in a specific order.
The requirement for informed consent can be waived if there is no ability to get the consent of the subject (e.g., the subject is incompetent) and there is not enough time to get consent from a legally authorized representative. In this case, both the investigator AND a physician who is not otherwise participating in the investigation must certify in writing that
- The subject is confronted by a life-threatening situation necessitating the use of the test article;
- Informed consent cannot be obtained because of the inability to communicate with, or obtain legally effective consent from the subject;
- There is not sufficient time to obtain consent from the subject’s legally authorized representative; and
- No alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the subject’s life
This pathway provides a mechanism for verifying that the intended use meets criteria for providing documentation of correspondence with FDA, including the emergency IND approval, the expanded access protocol/treatment plan and the proposed informed consent document.
The IRB Chair/Designee, can review and acknowledge the single patient use. This pathway also allows for submission of follow up information on the status of the patient. The acknowledgment of the single patient use by the IRB Chair/Designee, should not be construed as IRB approval. Only proposals that undergo full IRB review can receive IRB approval.
In the event of a waiver of informed consent for an emergency use, the IRB Chair/Designee, will confirm that both the physician holding the emergency IND and a physician who is not otherwise participating in the emergency use have certified in writing all of the following:
- the patient is confronted by a life-threatening situation necessitating use of the test article; informed consent cannot be obtained because of an inability to communicate with, or obtain legally effective consent from, the patient;
- time is not sufficient to obtain consent from the patient’s legal representative;
- no alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the patient’s life;
If, in the physician’s opinion, there is not sufficient time to obtain an independent physician’s determination that the criteria are met, the physician holding the emergency IND should make the determination and subsequently obtain (within five working days) a review of his/her determination by a physician not participating in the emergency treatment.
| Action | Descriptions and Further Information |
|---|---|
| 1. Request Letter of Authorization |
|
| 2. Submit Form 3926 to FDA |
|
| 3. Obtain IRB Concurrence |
|
| 4. Obtain Informed Consent |
|
- The physician must determine that the probable risk to the person from the investigational drug is not greater than the probable risk from the disease or condition; and
- FDA must determine that the patient cannot obtain the drug under another IND or protocol.
FDA FORM 3296 [For Emergency & Non-Emergency) Individual IND to Request IRB Concurrence
Box 10b: Request for Authorization to Use Form FDA 3926 I request authorization to submit this Form FDA 3926 to comply with FDA’s requirements for an individual patient expanded access IND.
|
UVA Study teams are NOT required to go through CRConnect or Protocol Builder for single patient treatment submission.
If you submit Form FDA 3926 to the FDA and selected the box under the Field 10.b (request for authorization to use alternative IRB review procedures: IRB CONCURRENCE):
Send the following documentation to the UVA IRB via IRBHSR@virginia.edu
Subject line: Single Patient Treatment Use-IRB Concurrence [Indicate Emergency or Non- Emergency]
- Choose either Emergency Use or Non-Emergency Use for a Single Patient
1. Expanded Access - Single Patient Emergency Use of an unapproved Drug and Biologic
OR
- Form 3926 (FDA)-see Instructions for Completion >>>>>>>>>>>>>>>>>>>ANSWER #10b YES
- Letter of Authorization (LOA) from study drug manufacturer (sample template HERE if needed)
- FDA Authorization letter to proceed with single patient treatment
- Single Patient IND# (assigned by FDA)
- Study specific informed consent-See UVA Template
- Treatment Plan/Protocol
- Investigator Brochure (IB)
- If applicable, Contact Investigational Drug Services (IDS): to make them aware of the single patient IND treatment in the event additional precautions need to be in place with IDS. Provide a copy of the IB and protocol/treatment plan
FDA may permit an investigational drug to be used for treatment of a patient population smaller than that typical of a treatment IND or treatment protocol. An intermediate-size population IND has no fixed numerical requirement, but it is for more than one patient and is generally employed when the investigational drug is not actively being developed for marketing. In cases where FDA has received a significant number of requests for individual patient expanded access for the same use, a sponsor may be asked to consolidate expanded access under this category.
In addition to the criteria listed at the beginning of this section for all expanded access, the FDA must also determine that there is enough evidence that the drug is safe at the proposed dose and duration and there is at least preliminary evidence of effectiveness of the drug as a therapeutic option in the patient population. For more information about FDA requirements, please see 21 CFR 312.315.
Procedures for IRB submission of expanded access protocol for intermediate –size populations
Intermediate size expanded access protocols must be submitted through usual procedures through CRConnect and the UVA IRB-HSR and requires full IRB review and approval under FDA regulations. Please see the School of Medicine website for information about preparing an IND submission to FDA. Submit all documents for pre-review via CRConnect and Protocol Builder.
Expanded access protocols for large patient populations are also referred to as treatment IND or treatment protocols. This category is used for widespread treatment use of an investigational drug. A widespread treatment IND is typically used to provide access to a large population and is often used to bridge the gap between completion of clinical trials and marketing approval. In addition to the criteria listed at the beginning of this section for all expanded access, FDA must also determine that the drug is being investigated in a controlled trial under an IND to support a marketing application for the expanded access or all clinical trials of the drug have been completed, the sponsor is actively pursuing marketing for approval of the expanded access and there is sufficient data supporting safety and effectiveness of the drug for the expanded access.
Procedures for IRB submission of expanded access protocols for large patient populations
Expanded access protocols for large patient populations must be submitted per usual procedures through CRConnect and Protocol Builder through the UVA IRB-HSR and requires full IRB review and approval under FDA regulations. Please see the School of Medicine website for information about preparing an IND submission to FDA.